



В настоящее время, пожалуй, уже практически невозможно в нашем отраслевом ландшафте найти предприятия, которые не стремились бы перевести свои бизнес-процессы в цифровой вид или хотя бы не задумывались об этом. В 2022 году, согласно исследованию, проведенному подразделением ISPE ЕАЭС, 48% респондентов охарактеризовали свою ситуацию как частичную цифровизацию. Лидирующими направлениями такой цифровизации стали складские операции (WMS), мониторинг производственной среды (EMS), управление документацией ФСК (QMS), контроль качества и маркировка готовой продукции (Track & Trace). Впрочем, при этом порядка 40% респондентов отметили, что имеют на данный момент преимущественно бумажные системы. Этот факт формирует достаточно емкие резервы для совершенствования.
При этом отмечу, что один из очевиднейших центров временных и ресурсных затрат либо «поглощен» емким понятием ФСК, либо «упущен из виду» – управление валидационной документацией. А ведь это внушительный сегмент в деятельности современных фармпредприятий. При этом в свете современных требований управлять этим сегментом становится все сложнее. В частности, по мере утверждения приложения 15 GMP в ЕАЭС (в виде Решения № 65 ЕЭК) мы наконец-то гармонизировали свои подходы в части валидации процесса производства, где таковая перестала быть разовым событием и получила связность с этапом фармацевтической разработки. Это вполне логично, поскольку CQAs (critical quality attributes, критические показатели качества) и связанные с ними CPPs (critical process parameters, критические параметры процесса) определяются именно на этапе фармацевтической разработки.

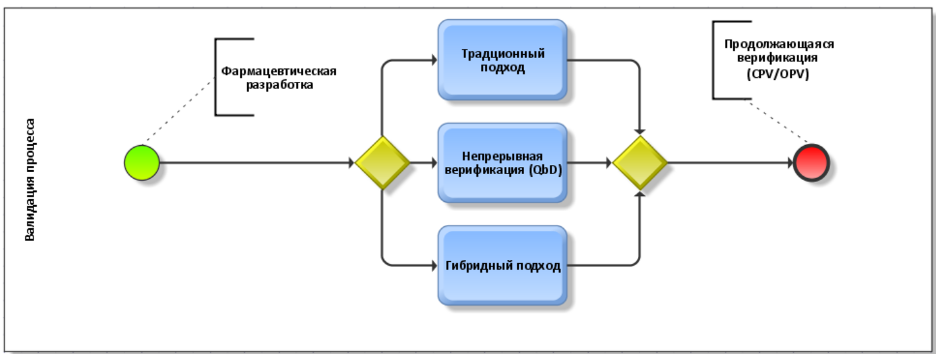
Адресуясь к рис. 1, отметим, что собственно валидация процесса производства согласно приложению 15 GMP может быть осуществлена тремя альтернативными способами – с реализацией традиционного подхода, путем непрерывной верификации (при условии использования подхода QbD – quality by design, качество через дизайн) или гибридного подхода, сочетающего в себе традиционный подход и QbD. Но, независимо от выбранного подхода, далее есть обязательство осуществлять продолжающуюся верификацию процесса в ходе жизненного цикла. Как нетрудно догадаться, реализация таких требований формирует большое количество документации, чья связность и непротиворечивость в бумажном виде весьма проблематична. Разумеется, что в данном случае цифровые решения читаются, что называется, «с порога».
Более того, даже в «обычной» квалификации помещений, оборудования и систем, где новшеств не так много, как может показаться на первый взгляд, тоже есть как большие объемы документации, так и запрос на связность решений. В частности, вторая редакция руководства ISPE Baseline 5: Commissioning & Qualification отмечает, что упомянутые помещения, оборудование и системы существует не в вакууме, а реализуют вполне конкретные технологии и предназначены для вполне конкретной номенклатуры продуктов. Для этих самых продуктов изначально должны быть определены CQAs, CPPs, что, в свою очередь, должно быть отражено в URS (user requirements specification, спецификация требований пользователя) на вновь создаваемые объекты квалификации и далее сформировать CAs (critical aspects, критические аспекты)/CDEs (critical design elements, критические элементы проекта), на основе которых впоследствии определяется объем квалификационных испытаний, формируются конкретные критерии приемлемости (см. рис. 2).

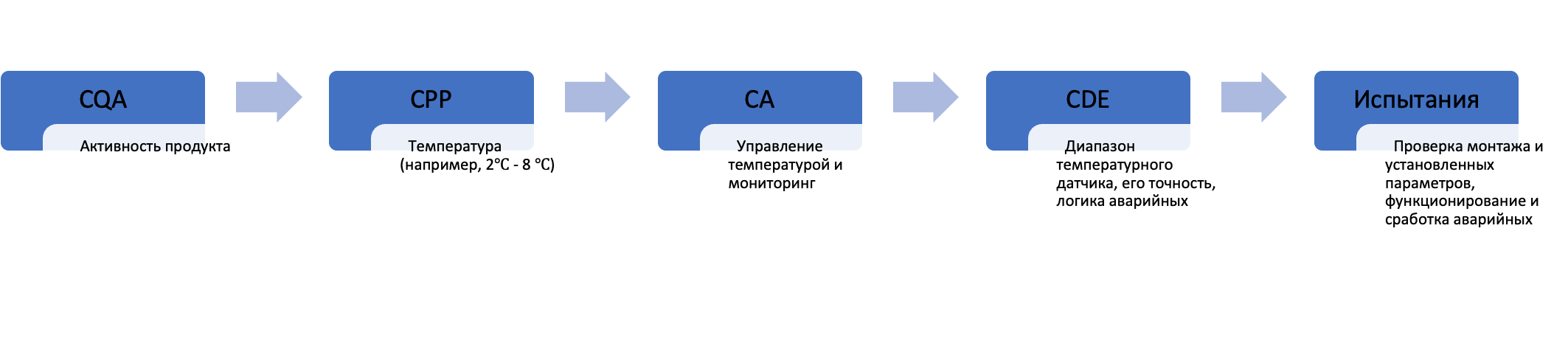
Если начать делать квалификацию в отрыве от конкретной технологии и номенклатуры продуктов, то могут случиться «разрывы функций». Например, если технология предусматривает работу с вязким продуктом, а система приготовления реализована для водных растворов, в результате чего не может быть осуществлено ни эффективное перемешивание, ни транспортировка продукта.
Другим важным аспектом квалификации является управление реквалификациями и пересмотрами, которое также должно быть организовано гибко. Гибкость эта заключается в том, что правила реквалификации, ее периодичность и объем не высечены в камне. Более того, собственно, реквалификация вообще является необязательной сущностью и чаще всего реализована для наиболее критичных объектов (чистых помещений, стерилизаторов и т. п.), а для широкого спектра менее критичных объектов предложен именно формат пересмотра, впрочем, с настройками переходов к различным уровням рассмотрения, вплоть до частичной или полной реквалификации, в зависимости от результатов пересмотра.
Разумеется, что заложить такие правила рациональнее всего в компьютеризированной системе, чтобы правила принятия решений были автоматизированы.

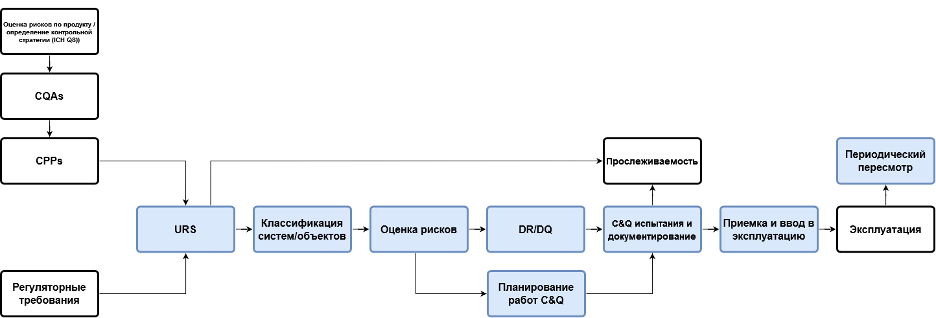
На рис. 3 представлена процессная карта, где проиллюстрированы функциональные блоки, между которыми должна быть организована взаимосвязь. Например, после URS предложена классификация объектов для того, чтобы выделить из их числа те, которые оказывают прямое влияние на качество продуктов и безопасность пациентов. «Полнометражная» квалификация требуется только для объектов, где такое прямое влияние определено. Для остальных единиц оборудования и систем следует руководствоваться принципами GEP (ISPE Good Practice Guide: Good Engineering Practice 2nd Edition). Как можно реализовать такую классификацию объектов? Путем ответов на следующие вопросы:
| Номер | Критериальный вопрос оценки влияния |
| Q1 | Содержит ли система CAs/CDEs или выполняет функции, которые способствуют реализации одного или нескольких требований к процессу (CQAs), включая CPPs? Дополнительно прокомментируйте, если положительный ответ является результатом взаимодействия с системой управления/автоматизации в пределах оцениваемой системы. |
| Q2 | Имеет ли система прямой контакт с продуктом или процессным потоком и имеет ли такой контакт потенциальное влияние на качество готовой продукции или являет собой риск для пациента? |
| Q3 | Предоставляет ли система вспомогательные компоненты или производит ингредиенты или растворители (например, ВДИ) и может ли качество (и, таким образом, соответствие спецификациями) таких субстанций повлиять на качество готовой продукции или являть собой риск для пациента? |
| Q4 | Используется ли система для очистки, санитизации, стерилизации и может ли ее неисправность результировать в несоответствующую очистку, санитизацию или стерилизацию, таким образом формируя риск для пациента? |
| Q5 | Предоставляет ли система требуемую среду (например, защитный слой азота, закрытые процессы, воздух для зоны наполнения соответствующего качества, поддержание температуры, влажности, когда такие параметры являются частью CPPs для продукта) для процесса и может ли неисправность таких систем формировать риск для пациента? |
| Q6 | Используется ли система для создания, обработки и сохранения данных, используемых для выпуска или забраковки продукции, CPPs, или электронных записей, являющихся предметом 21 CFR Part 11 и приложения 11 GMP, или эквивалентных локальных норм? |
| Q7 | Укупоривает ли система контейнеры или защищает продукт, сбой которой являет собой риск для пациента или может вызвать деградацию качества продукта? |
| Q8 | Предоставляет ли система идентификационную информацию в отношении продукта (например, номер серии, срок годности, противоконтрафактные меры) без независимой верификации или используется для верификации такой информации? |
Если хотя бы на один из таких вопросов получен положительный ответ, то система считается таковой, которая оказывает прямое влияние. Для таких систем следует проводить дальнейшую оценку рисков и в соответствии с такой оценкой формировать объем квалификационных испытаний и формулировать критерии приемлемости. При этом перечень объектов квалификации на основании ответов на вопросы будет сформирован в рамках компьютеризированной системы автоматически в план-график, который является приложением к валидационному мастер-плану (ВМП). При этом, как показано на рис. 3, важна прослеживаемость требований. Компьютеризированная система в состоянии сформировать матрицу прослеживаемости требований в автоматизированном режиме, обеспечив связь начиная с URS вплоть до отчетов о квалификации.
После завершения первичной квалификации на основании оценки рисков определяется вид последующих работ (реквалификация или пересмотр) и их периодичность. Одним из вариантов такой реализации может являться итерационный подход на основании методологии FME(C)A, когда мы оперируем тяжестью возможных последствий отказа (S = severity), вероятностями возникновения (O = occurrence) и обнаружения (D = detectability) таких отказов. Эти категории позволяют нам рассчитать приоритет риска RPN ( = S × O × D) по каждой из рисковых позиций. Можно настроить пороговые уровни, которые адресуют нас к ежегодным пересмотрам определенных единиц оборудования и/или линий, пересмотрам раз в два года и т. п.
Но, пожалуй, главным преимуществом электронной системы для управления валидацией является возможность перейти на полностью безбумажный процесс (paperless validation), только посредством реализации электронных записей и электронных подписей. Причем последнее – это, в общем-то, свежайший оформившийся отраслевой тренд в международном масштабе, недаром статья в последнем номере журнала ISPE Pharmaceutical Engineering выделяет этот вопрос отдельно.
Многие годы непосредственной деятельности автора в «полях», на квалификационных испытаниях, свидетельствуют, что хоть в GMP и отсутствует понятие «черновик», но я многократно наблюдал, как вносятся записи в формы протоколов результата, как в итоге зарисовываются схемы на произвольных листах А4 или в блокноты (в т. ч. представителями международной фармы, сотрудниками транснациональных корпораций). Почему так? В «полях», в чистых помещениях, зачастую условия далеки от офисных, и, например, можно набросать определенный комментарий даже в специально отведенном поле заполняемого бланка, но сделано это может быть неидеально с эстетической точки зрения, или поля, предусмотренного протоколом квалификации для ввода комментария, недостаточно, и мы начинаем делать «пометки» с обратной стороны листа заполняемого бланка и выписывать тому подобные пируэты. В результате в офисе происходит «перезаполнение» таких бланков, что само по себе – насмешка над принципами целостности данных. Разумеется, выйдя в «поля» с планшетом, как минимум почерк нас перестанет смущать – комментарий будет выглядеть одинаково, независимо от места заполнения, недостаточность места в поле для комментария может быть сразу же устранена путем генерации дополнительного бланка (например, создаваемого по предопределенному правилу с предопределенными метаданными вида <номер бланка>.1-<код протокола>) контролируемым образом, с фиксацией в аудиторском (контрольном) следе и без диссонанса в части эстетики. Не говоря уже о том, что, проставив заключения по испытаниям в электронном виде во всех заполняемых бланках, компьютеризированная система впоследствии уже сама вполне сможет предложить сводную таблицу результатов испытаний для отчета с переносом в него требуемых данных, устраняя человеческий фактор при ручном переносе и высвобождая ресурс сотрудников для других задач.
Востребованность таких решений очевидна, тем более что опыт «систем-пионеров» в этом плане – ЛИМС, СЭД и т. п. – вполне является позитивным, ведь идеологически они реализуют сходные цели. Та же система ЛИМС позволяет «не потерять пробы на подоконниках» в лабораториях ОКК. СЭД позволяет перестать носиться с кипой бумаг и «расписывать ручки» в листах согласования и обучения. Валидационный фланг также ожидает своего часа.
Конечно, скептики могут возразить: мол, препятствием к внедрению таких систем является исчезновение «пространства для маневра» – нельзя будет что-то внести задним числом, нельзя будет «перескочить» через зафиксированное отклонение и т. п. Но, во-первых, в подавляющем большинстве случаев это следствие неоптимальных бизнес-процессов (просто бумажные системы это не подсвечивали столь контрастно), во-вторых, те же «препятствия» существуют и для других компьютеризированных систем, что, однако, отнюдь не воспрепятствовало их внедрению. Например, CDS (chromatograph data system, хроматографическая система данных) очень даже зафиксирует, не выбрали ли мы из семи результатов три «лучших», – тем не менее используются такие системы повсеместно – и уже много лет. Они, собственно, и создавались отчасти для того, чтобы сделать такие манипуляции невозможными. Далее эти системы могут передавать данные в ЛИМС, где будет автоматизированно формироваться аналитический лист или паспорт готовой продукции.
В части квалификации и валидации можно сформулировать, что в ряде случаев, напротив, очень важно не потерять информацию, скажем, о не прошедших испытание на целостность НЕРА-фильтрах. Это означает, что в определенную дату будет заполнен бланк, фиксирующий отрицательный результат, и, скажем, на следующей день будет осуществлена замена НЕРА-фильтров и повторное тестирование. Этого не нужно опасаться и пытаться как-то «избегать». Во-первых, такой факт нужен, как правило, инженерно-техническому флангу предприятия для учета замены фильтров (иначе не вполне понятно, почему идет расход недешевого запаса и соответствующий новый заказ), во-вторых, сам факт наличия нецелостного НЕРА-фильтра вовсе не означает, что класс помещения не будет поддерживаться, во всяком случае для классов D, C, B однозначно локальная утечка погоды не сделает, если фильтр «не проткнули вилами». Регуляторы также прекрасны знакомы с этой спецификой, и вопросы вызовет скорее «стерильная» чистота в записях в части целостности НЕРА-фильтров на протяжении многих лет для одних и тех же объектов.
Таким образом, можно заключить, что вопрос состоит в том, как именно будут реализовываться подобные системы, какие функции будут автоматизированы (формирование и отслеживание планов-графиков, оценка рисков), будут ли в них внедрены элементы поддержки принятия решений (анализ заполненных бланков с выдачей автоматизированного заключения и т. п.). «Теневые MS Excel» и бумажные бланки, скорее всего, в ближайшее время канут в Лету, хотя и останутся номинально разрешены (для MS Excel, разумеется, при условии вывода из «тени» посредством той же валидации).
Завершу повествование достаточно известной историей, когда математик задал вопрос гуманитарию: мол, в задаче известно, что озеро зарастает ряской целиком за 30 дней, а также известно, что ежедневно количество ряски на поверхности озера удваивается. Вопрос: за какое количество дней озеро зарастет ряской наполовину? Гуманитарий поспешно отвечает: за 15 дней, хотя правильный вариант ответа – 29 дней. Это пример геометрической прогрессии. Т. е. последнее удвоение произошло в последний, 30-й день. А за два дня всего четверть озера была покрыта ряской, за три дня – 1/8, за четыре – 1/16. Т. е. много дней уже шел процесс, который мало кто замечал, и только в последние дни эффект стал очевидным для всех. На этом фоне ИТ-решения развиваются даже не в геометрической прогрессии, а по экспоненте. А вслед за ними подтягивается и нормативно-рекомендательная база – обновленное руководство ISPE GAMP 5, проект FDA в части CSA (computer software assurance), концептуальная статья PIC/S в части обновления приложения 11 GMP (посвященного компьютеризированным системам и по новому концепту допускающего использование инноваций в фарма- Agile, CSA etc.). Sapienti sat.