
Обновленное Приложение 15 GMP ЕАЭС (в форме Решения Совета ЕЭК от 14 июля 2021 года № 65) официально закрепило возможность применения подхода Quality by Design – «качество через дизайн». Вместе с тем, если пойти немного вглубь, то в рамках ЕАЭС концепция «качество через дизайн» впервые прозвучала еще в тексте Рекомендации Коллегии ЕЭК от 26 сентября 2017 года № 19. По сути, и Решение № 65 (соответствующее в целом Приложению 15 GMP EU), и Рекомендация № 19 (соответствующая в целом Руководству ЕМА по валидации процесса) всего лишь актуализируют достаточно давно прорабатываемый концепт, сменяющий подход Quality by Testing, как неприемлемый ввиду запредельной контрпродуктивности. Тестирование на финальном этапе имеет значительный риск забраковать продукцию, на производство которой уже затрачены немалые ресурсы.
Что же скрывается за словосочетанием «качество через дизайн» (Quality by Design), которое теперь предложено к рассмотрению? Оттолкнемся от определения, данного по текстам Решения № 65 и Рекомендации № 19.
«Качество через дизайн» (Quality by Design) – это системный подход, предусматривающий определение целей перед началом разработки продукции, точную оценку продукции и процесса ее производства, а также контроль процесса производства на основе научных данных и принципов управления рисками для качества.
Звучит как «пугающее наукообразие». Мало того что валидация процесса на современном этапе перестает быть разовым событием (уже исключен вариант произвести «кое-как» три валидационные серии и «забыть» о валидации на какое-то время), так вдобавок еще и предлагается осуществлять контроль процесса производства на основе научных данных. То есть наукоемкость и ресурсоемкость процесса очевидно возрастают. При этом не следует забывать, что если подход «качество через дизайн» является в настоящее время допустимой альтернативой, то продолжающаяся верификация процесса в ходе жизненного цикла становится безальтернативным обязательством.
Казалось бы, эти нововведения могут восприниматься как очередной «регуляторный прессинг», «гиря на ногах бизнеса». Однако при более детальном рассмотрении выясняется, что как раз реализация концепта «качество через дизайн» способна сократить затраты на выведение на рынок новых лекарственных препаратов вплоть до 25%. За счет чего может быть достигнут такой эффект? По сравнению с традиционным подходом, когда этап фармразработки выполнялся без должного статистического обоснования, во многом эмпирическим путем, «по наитию», больший фокус внимания уделяется именно этапу фармацевтической разработки – именно там формируется целевой профиль продукта, связанные с ним CQA (критические показатели качества), CPP (критические параметры процесса), как возможная опция – пространство рабочих параметров (design space) и стратегия контроля. Считается, что реализация такого концепта не принесет значимой экономии на этапе фармразработки, однако обеспечит статистически обоснованные знания о продукте и процессе, что заметно упростит масштабирование, трансфер и производство последующих коммерческих серий, поскольку процесс будет заведомо в состоянии статистического контроля (см. рис. 1).
{kind=link}
Верификация стратегии контроля фармразработки
При реализации концепта «качество через дизайн» и, соответственно, выработке стратегии контроля еще на этапе фармразработки далее речь идет о верификации этой самой стратегии. В Приложении № 15 это сформулировано как непрерывная верификация процесса, переходящая в продолжающуюся верификацию процесса в ходе жизненного цикла. Опциональное применение пространства проектных параметров (design space) имеет еще одну существенную выгоду – варьирование параметров в рамках определенного проектного поля может не рассматриваться как изменение. Это в итоге минимизирует затраты ресурсов как при рассмотрении таких вариаций в рамках процедур управления изменений на предприятии, так и с точки зрения внесения потенциальных изменений в регистрационное досье на препарат, а это уже заметное преимущество. Перерегистрация и внесение изменений в регистрационное досье – процесс, как известно, не быстрый.
В целом жизненный цикл продукта условно разбивается на три стадии (см. рис. 2).
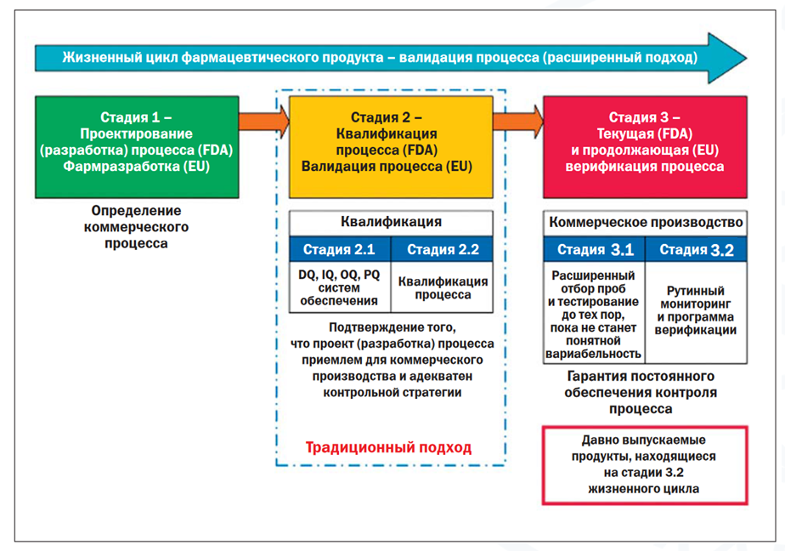{kind=link}
Ввиду того что валидация процесса перестает быть разовым событием, возрастает важность внятно определенных CQA, CPP и верно выработанной стратегии контроля, отражающей особенности процесса. Ведь если это не так, то постоянная фиксация межсерийной вариабельности будет обузой во всех смыслах, влекущей за собой производственные и регуляторные риски, перегруженность лабораторий контроля качества дорогостоящим подтверждением вариабельности по непродуманным стратегиям контроля с риском последующих итераций. Поэтому единственным разумным вариантом видится перестать «строить дом с крыши» и с самого начала продумать процесс, так сказать, с «фундамента».
Что делать фармпроизводителям, чьи препараты уже реализуются на рынке?
Вместе с тем руководства ISPE, в частности, рассматривают и случаи, когда продукт уже находится в коммерческой реализации – т. н. legacy products. В этом случае продукция начинает свой жизненный цикл с этапа 3.2 (см. рис. 2). Это особенно важный аспект в свете того, что новая версия Приложения № 15 в ЕАЭС вступила в силу только с февраля 2022 года, а до этого момента, безусловно, в коммерческой реализации находилось большое количество уже зарегистрированных лекарственных препаратов, чья первичная валидация уже была проведена до этого. Для таких препаратов необходимо реализовать продолжающуюся верификацию процесса в ходе жизненного цикла, первично отталкиваясь от данных производства, устанавливая CQA, CPP (если этого не было сделано ранее) и в дальнейшем при необходимости внося изменения и/или дополнения в планы отбора проб с целью статистического контроля процесса производства.
Из рациональных предложений в этом плане можно выделить то, что такая деятельность станет частью привычных регулярных обзоров качества продукции с тем акцентом, что будет обеспечена фиксация событий в режиме реального времени (а не ретроспективно), это позволит при необходимости реализовывать своевременные САРА. Разумеется, в ходе годовых обзоров качества не нужно будет дублировать эту деятельность, а можно просто сослаться на уже построенные тренды и их анализ.
В ходе такой продолжающейся верификации процесса будут возрастать знания в отношении самого процесса, также будут установлены источники возможной вариабельности. При необходимости могут быть реализованы изменения, возвращающие процесс как на вторую стадию (повторную валидацию при внесении изменений), так и на первую (повторную фармразработку).
Для новых лекарственных продуктов настоятельно рекомендуется использовать подход «качество через дизайн», что призвано значительно снизить вероятность таких «возвратов» на первой и второй стадиях, что, несомненно, является прямой потерей времени и ресурсов. И пусть фармсообщество не вводит в заблуждение тот факт, что этот подход – «всего лишь» альтернатива «традиционному подходу». Метод проб и ошибок – достаточно дорогостоящий. Именно исключение таких непроизводительных затрат времени и ресурсов является ключевым выходом и ключевой выгодой при следовании подходу «качество через дизайн». Ведь вся суть такого подхода – это статистически значимая заявка на успех еще на этапе фармразработки.
Как известно, валидация в целом (как система) и валидация процессов (как ключевой элемент этой системы) являются базисами построения и поддержания фармацевтической системы качества, позволяющими с определённым уровнем «уверенности» гарантировать качество производимой лекарственной продукции в ходе её жизненного цикла.
Однако, безусловно, необходимо поэтапно совершенствовать и обновлять в т. ч. сами подходы и методологию валидации процессов. И здесь важно также придерживаться принципов международной гармонизации в части нормативно-технических требований к производству лекарственных средств, что, собственно, и стараются делать не только национальные и наднациональные регуляторы, но и некоммерческие структуры, как, например, ISPE ЕАЭС и Евразийская Академия надлежащих практик, создавая обучающие продукты для фармпроизводителей ЕАЭС, посвященные валидации технологического процесса на основании жизненного цикла продукта.